[CROI 2013]M.Stevenson: Reservoir, persistenza virale e cura
Inviato: lunedì 4 marzo 2013, 13:53
Mario Stevenson, University of Miami, ha tenuto ieri una lunga ma molto chiara lezione ai giovani ricercatori, che funge da introduzione allo stato dell’arte della ricerca su reservoir, persistenza virale e speranze di una cura.
Research on Viral Reservoirs, Persistence, and Cure

Sappiamo che la ART funziona e supporta un ambiente aviremico per lunghi periodi di tempo.
Ma in ogni individuo che sospende la terapia si verifica un rapido rebound virale, indipendentemente dalla lunghezza del periodo aviremico.
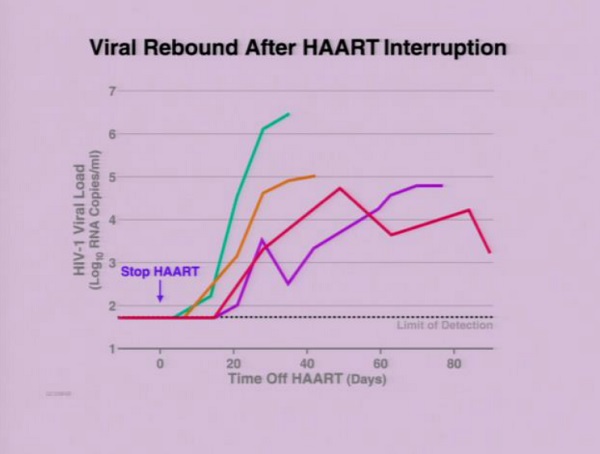
Quindi l’unica cosa che è davvero necessario fare è stanare l’HIV da qualunque parte del corpo riesca a nascondersi.
Ma è un compito che si sta rivelando più difficile di quanto si pensasse in passato. Inoltre, la discussione su che cosa supporti la persistenza virale a fronte di una ART che funziona è più aperta che mai.

Riguardo alla persistenza virale, esistono due ipotesi – forse non mutualmente esclusive:
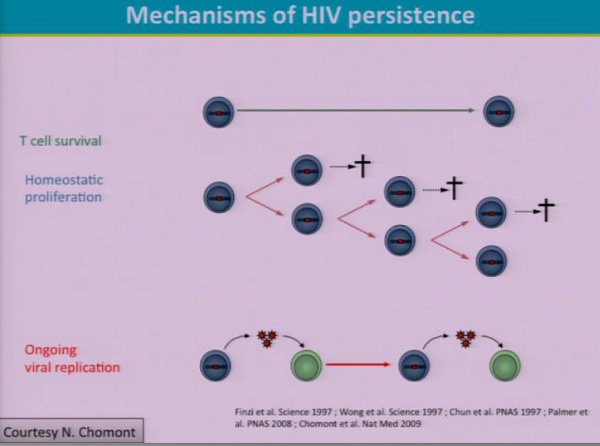
Nell’ultimo paio di anni, ci sono state grandi discussioni sul ruolo della replicazione residua attiva nel mantenere il reservoir latente o nel mantenere la persistenza virale.
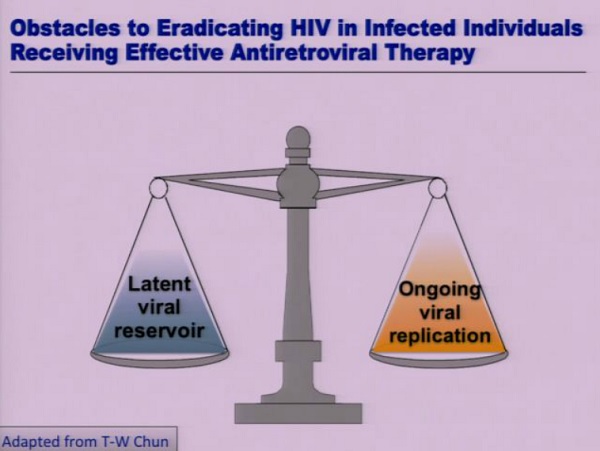
Esistono un paio di regole di base riguardo a ciascuna delle ipotesi e, soprattutto, un paio di importanti predizioni basate su queste ipotesi.
Nel caso della latenza, la premessa è che la terapia antiretrovirale blocca ogni replicazione virale e, in una situazione in cui non c’è replicazione attiva, la stabilità intrinseca del reservoir latente è dovuta alla longevità dei CD4 quiescenti, che conferiscono memoria immunologica al sistema immunitario.
La predizione importante è che se si prova ad intensificare la terapia antiretrovirale ci si deve aspettare di non vedere alcun impatto sul reservoir.

L’altra ipotesi è che durante il trattamento antiretrovirale non si riesca a sopprimere completamente la replicazione del virus e che il reservoir venga ricaricato da infezioni de novo.
La predizione importante, in questo caso, è che l’intensificazione terapeutica possa avere qualche effetto sul reservoir.

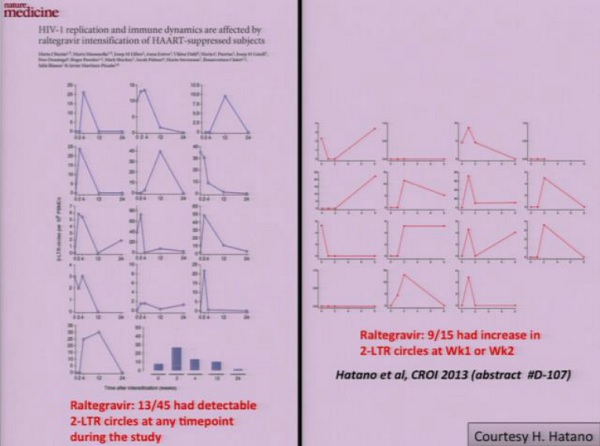
Ecco perché al CROI, nella sezione dedicata all’eradicazione, viene presentato da Hiroyu Hatano uno studio clinico sull’intensificazione della ART mediante Raltegravir, che è molto simile – nel suo impianto – allo studio di Maria Buzon e dal gruppo di Barcellona pubblicato pochi anni fa.

Al centro di questo genere di studi sono i circoli 2-LTR di DNA virale integrato, tipici dei retrovirus, con il Raltegravir che, in caso di infezioni de novo, dovrebbe interferire con l’integrazione del virus nel genoma.
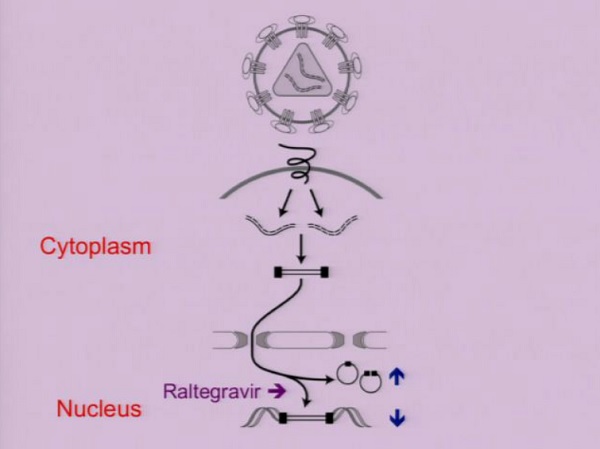
E così parrebbe, guardando i risultati appaiati dei due diversi studi.
Come interpretare tutto questo?
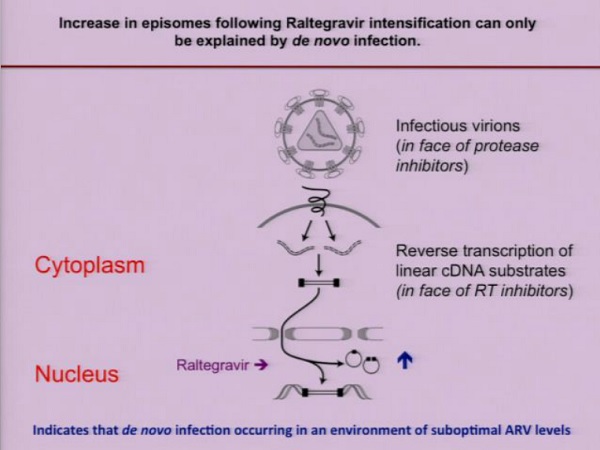
A seguito di intensificazione con Raltegravir, si ha un aumento degli episomi e questo può significare soltanto che c’è stata una infezione de novo e che, quindi, la capacità della ART di bloccare la replicazione non è ottimale.
E tuttavia in quasi tutti gli studi fatti finora non si è visto

Allora, che cosa potrebbe riconciliare risultati e osservazioni apparentemente così contrastanti?
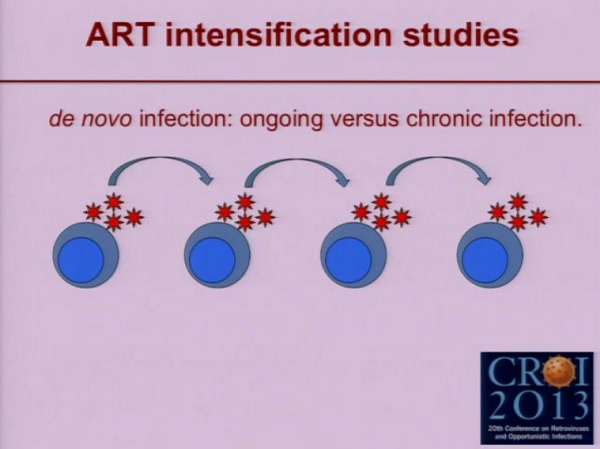
Il termine “replicazione attiva” [ongoing replication] viene usato in due accezioni diverse: i virologi pensano a una cellula che viene infettata e poi trasmette l’infezione ad un’altra, e così via. Questa è un’infezione attiva che si diffonde.
Una spiegazione alternativa, che potrebbe permettere di riconciliare alcune di queste osservazioni così in contrasto, è che esista una fonte di infezione cronica che scatena infezioni de novo e che questo accada in presenza di ART soppressiva.
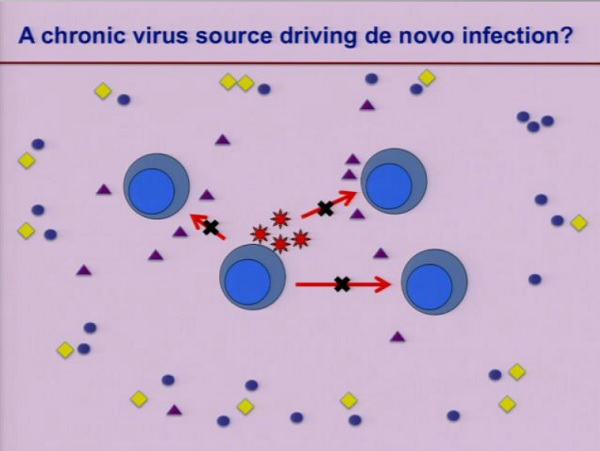
Questo significa che, nei comparti in cui si verificano queste infezioni de novo, la penetrazione degli ARV è subottimale e quindi che l’intensificazione con Raltegravir può arrivare dove una ART normale non arriva, impedendo l’innesco di nuovi cicli di infezione.

È possibile dunque che il Raltegravir sia una sorta di Heineken della ART, perché riesce a raggiungere dei comparti meno facilmente raggiungibili da altri antiretrovirali.
Ma questa infezione residua, de novo, è rilevante da un punto di vista clinico?
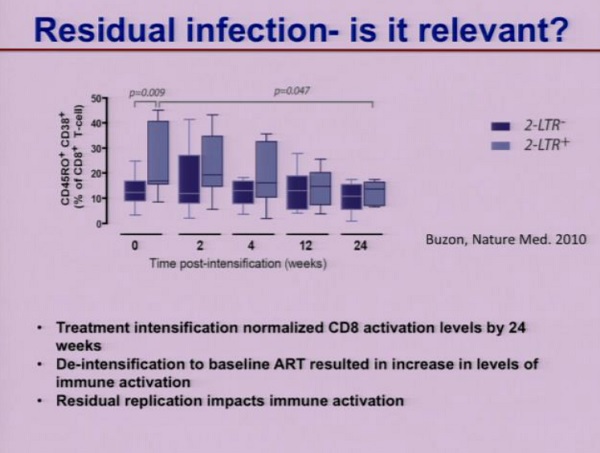
Lo studio di Maria Buzon del 2010 ha dimostrato che persone che avevano più alti livelli di partenza di infezione residua avevano anche livelli più alti di CD8 attivati e che questi si normalizzavano dopo 24 settimane di intensificazione, mentre il ritorno alla ART normale comportava un aumento dei livelli di attivazione immunitaria. Dunque che LA REPLICAZIONE RESIDUA HA UN QUALCHE TIPO DI EFFETTO SULL’ATTIVAZIONE IMMUNITARIA.
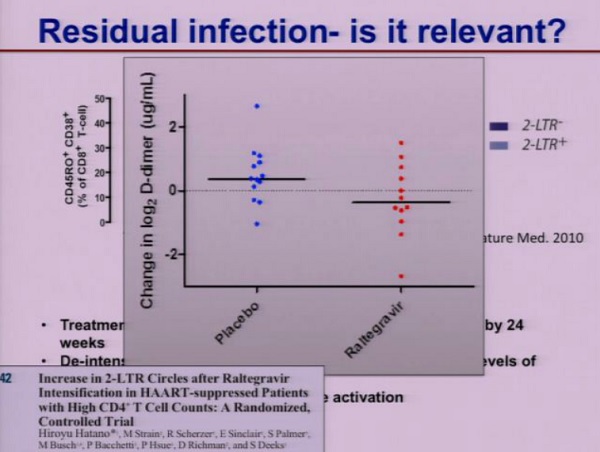
Nello studio di Hatano che viene presentato a questo CROI quello che si vede è che le persone che hanno intensificato la terapia con Raltegravir hanno avuto una riduzione significativa dei livelli di D-dimero, che è un marker molto importante di rischio cardiovascolare.
Questo fa dunque ipotizzare che la replicazione virale residua ABBIA un qualche significato clinico.
L’idea che sta dietro la DE-INTENSIFICAZIONE della ART è di eliminare della terapia antiretrovirale, in modo strutturato e sicuro, per arrivare a capire qual è la fonte della recrudescenza virale e quale la natura dei reservoir. Di qui alcuni studi che verranno presentati in questi giorni.

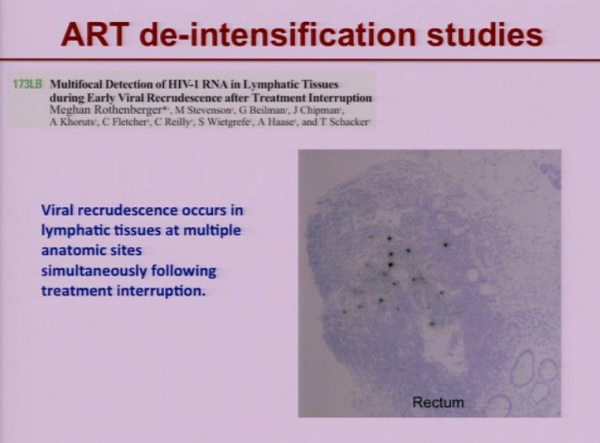
Lo studio di Schacker dimostra che , dopo interruzione del trattamento, si ha un rebound virale nei tessuti linfatici, simultaneamente in diversi comparti anatomici.
Questo può dipendere o dalla presenza di un grande serbatoio di cellule latentemente infette – e allora ci si deve chiedere come abbiano fatto ad attivarsi tutte insieme in diversi siti anatomici.
Oppure (spiegazione alternativa, ma non necessariamente incompatibile) da cellule che sono infette non in modo latente, ma produttivo, che non sono in grado di causare nuove infezione finché subiscono la pressione della ART ma, quando questa viene sospesa, possono innescare nuovi cicli di infezione.

Passiamo ora alle diverse strategie che la ricerca sta mettendo in atto per arrivare all’eradicazione.
Una strategia che è già passata in fase clinica è lo “shock and kill” – il tentativo di ripulire il reservoir attivando il virus dalla latenza.
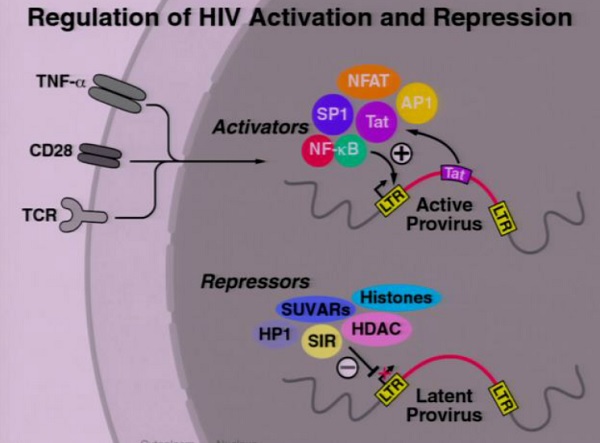
Attivatori della latenza dell’HIV.
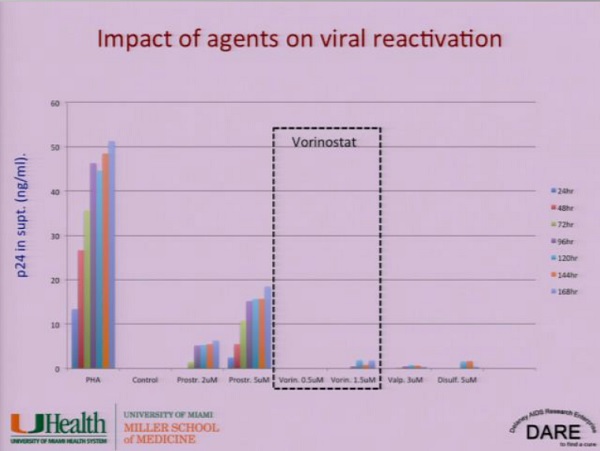
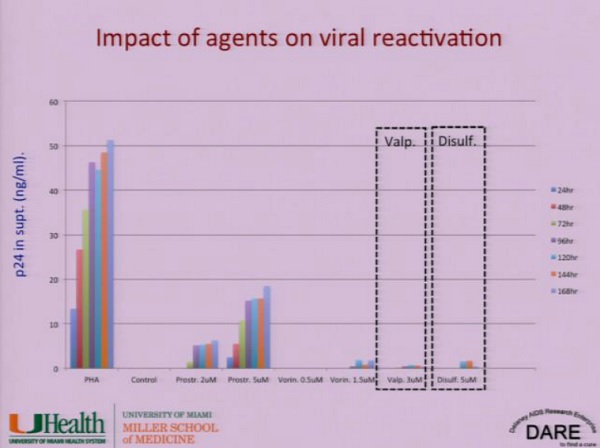
Il farmaco antilatenza che è stato ad oggi più studiato è il Vorinostat, con risultati per ora assai ambigui. [NdD: è vero che lo studio portato al congresso dalla Lewin riguarda soprattutto la sicurezza della somministrazione di questo HDACi ma … che delusione!!]
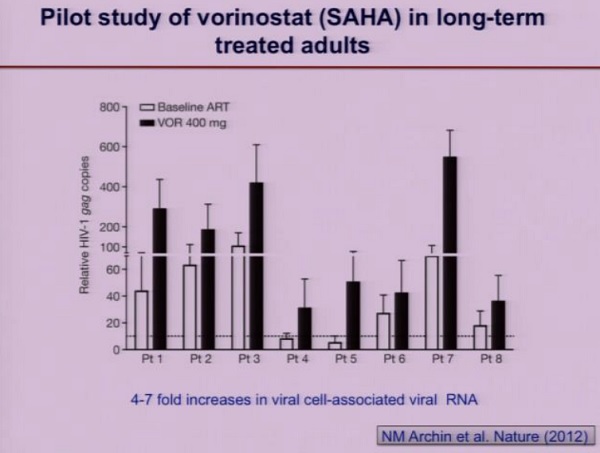
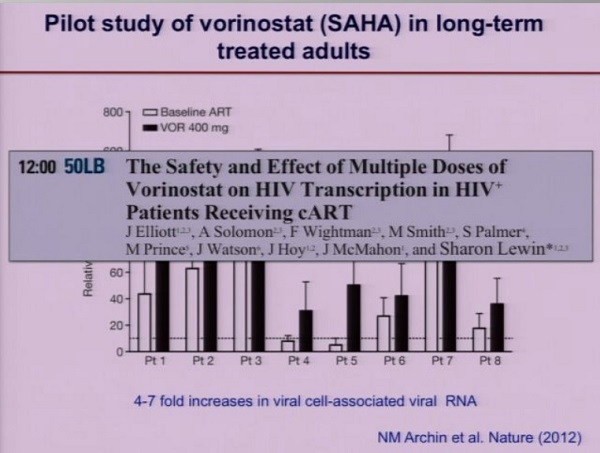
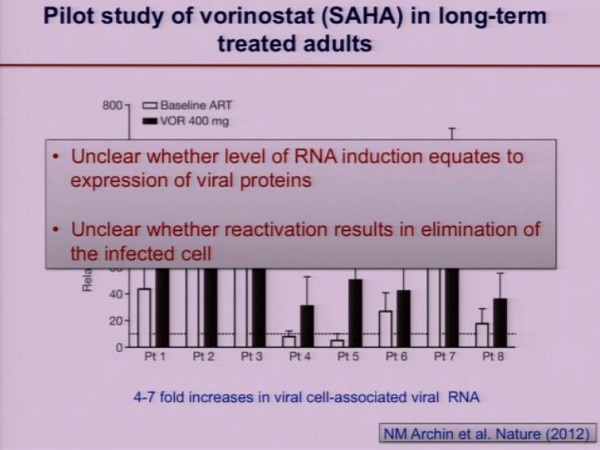
Le aspettative di qualsiasi strategia di “purging” dei reservoir sono che la riattivazione del virus porti all’eliminazione delle cellule infette, sia grazie agli aspetti citopatici del virus, sia grazie alla reazione immune dell’ospite.
Ma la strada è resa più difficile dal fatto che Siliciano ha dimostrato che la frequenza di provirus capaci di replicazione è più alta di quanto si pensasse.

C’è poi la questione dei reservoir nelle cellule della linea mieloide, e segnatamente i macrofagi, che potrebbero – se si dimostrassero davvero un reservoir presente in persone con viremia soppressa dalla ART - richiedere delle strategie di eradicazione diverse da quelle che si stanno sperimentando contro cellule della linea linfoide.

La ricerca che sta facendo Stevens riguarda proprio l’uso di Vorinostat, acido valproico e Disulfiram su linee cellulari di macrofagi.
In conclusione, le questioni principali che rimangono aperte riguardano

Alla fine della lezione, rispondendo a una domanda sulle prospettive dei farmaci antilatenza da qui a 5 anni, Stevenson ha risposto che ha dei grossi dubbi sui test che abbiamo oggi a disposizione per studiare gli effetti di queste sostanze sulla latenza virale. Il gold standard continua ad essere la rilevazione di virus capace di replicazione, ma i test oggi correnti mancano di precisione e danno risultati che non si riescono a riprodurre da un laboratorio all'altro [cosa che è emersa in modo INCREDIBILE l'estate scorsa, quando non si capiva se fosse stato trovato o no del virus capace di replicarsi in Timothy Brown].
Altra domanda: come combinare le diverse strategie per attaccare i reservoir?
Risposta: unire a una strategia antilatenza la soluzione del "problema di Shen-Siliciano" e fare in modo di rinforzare gli effetti citotossici dei CD8 [vabbè, questo l'avevamo capito anche noi fin dall'anno scorso ...]
Research on Viral Reservoirs, Persistence, and Cure
Sappiamo che la ART funziona e supporta un ambiente aviremico per lunghi periodi di tempo.
Ma in ogni individuo che sospende la terapia si verifica un rapido rebound virale, indipendentemente dalla lunghezza del periodo aviremico.
Quindi l’unica cosa che è davvero necessario fare è stanare l’HIV da qualunque parte del corpo riesca a nascondersi.
Ma è un compito che si sta rivelando più difficile di quanto si pensasse in passato. Inoltre, la discussione su che cosa supporti la persistenza virale a fronte di una ART che funziona è più aperta che mai.
Riguardo alla persistenza virale, esistono due ipotesi – forse non mutualmente esclusive:
- 1) la longevità dei reservoir virali che sosstengono l’HIV in presenza di ART è dovuta alla stabilità intrinseca dei CD4 latentemente infetti, con la proliferazione omeostatica, il meccanismo ipotizzato da Nicholas Chomont per spiegare il perpetuarsi del reservoir;
2) un’ipotesi meno popolare è quella di una sorta di replicazione virale che si mantiene anche durante la ART.
Nell’ultimo paio di anni, ci sono state grandi discussioni sul ruolo della replicazione residua attiva nel mantenere il reservoir latente o nel mantenere la persistenza virale.
Esistono un paio di regole di base riguardo a ciascuna delle ipotesi e, soprattutto, un paio di importanti predizioni basate su queste ipotesi.
Nel caso della latenza, la premessa è che la terapia antiretrovirale blocca ogni replicazione virale e, in una situazione in cui non c’è replicazione attiva, la stabilità intrinseca del reservoir latente è dovuta alla longevità dei CD4 quiescenti, che conferiscono memoria immunologica al sistema immunitario.
La predizione importante è che se si prova ad intensificare la terapia antiretrovirale ci si deve aspettare di non vedere alcun impatto sul reservoir.
L’altra ipotesi è che durante il trattamento antiretrovirale non si riesca a sopprimere completamente la replicazione del virus e che il reservoir venga ricaricato da infezioni de novo.
La predizione importante, in questo caso, è che l’intensificazione terapeutica possa avere qualche effetto sul reservoir.
Ecco perché al CROI, nella sezione dedicata all’eradicazione, viene presentato da Hiroyu Hatano uno studio clinico sull’intensificazione della ART mediante Raltegravir, che è molto simile – nel suo impianto – allo studio di Maria Buzon e dal gruppo di Barcellona pubblicato pochi anni fa.
Al centro di questo genere di studi sono i circoli 2-LTR di DNA virale integrato, tipici dei retrovirus, con il Raltegravir che, in caso di infezioni de novo, dovrebbe interferire con l’integrazione del virus nel genoma.
E così parrebbe, guardando i risultati appaiati dei due diversi studi.
Come interpretare tutto questo?
A seguito di intensificazione con Raltegravir, si ha un aumento degli episomi e questo può significare soltanto che c’è stata una infezione de novo e che, quindi, la capacità della ART di bloccare la replicazione non è ottimale.
E tuttavia in quasi tutti gli studi fatti finora non si è visto
- 1) alcun impatto dell’intensificazione della ART sulla viremia residua;
2) né sui livelli di DNA virale;
3) né sull’RNA associato alle cellule;
4) né sull’evoluzione delle sequenze virali.
Allora, che cosa potrebbe riconciliare risultati e osservazioni apparentemente così contrastanti?
Il termine “replicazione attiva” [ongoing replication] viene usato in due accezioni diverse: i virologi pensano a una cellula che viene infettata e poi trasmette l’infezione ad un’altra, e così via. Questa è un’infezione attiva che si diffonde.
Una spiegazione alternativa, che potrebbe permettere di riconciliare alcune di queste osservazioni così in contrasto, è che esista una fonte di infezione cronica che scatena infezioni de novo e che questo accada in presenza di ART soppressiva.
Questo significa che, nei comparti in cui si verificano queste infezioni de novo, la penetrazione degli ARV è subottimale e quindi che l’intensificazione con Raltegravir può arrivare dove una ART normale non arriva, impedendo l’innesco di nuovi cicli di infezione.
È possibile dunque che il Raltegravir sia una sorta di Heineken della ART, perché riesce a raggiungere dei comparti meno facilmente raggiungibili da altri antiretrovirali.
Ma questa infezione residua, de novo, è rilevante da un punto di vista clinico?
Lo studio di Maria Buzon del 2010 ha dimostrato che persone che avevano più alti livelli di partenza di infezione residua avevano anche livelli più alti di CD8 attivati e che questi si normalizzavano dopo 24 settimane di intensificazione, mentre il ritorno alla ART normale comportava un aumento dei livelli di attivazione immunitaria. Dunque che LA REPLICAZIONE RESIDUA HA UN QUALCHE TIPO DI EFFETTO SULL’ATTIVAZIONE IMMUNITARIA.
Nello studio di Hatano che viene presentato a questo CROI quello che si vede è che le persone che hanno intensificato la terapia con Raltegravir hanno avuto una riduzione significativa dei livelli di D-dimero, che è un marker molto importante di rischio cardiovascolare.
Questo fa dunque ipotizzare che la replicazione virale residua ABBIA un qualche significato clinico.
L’idea che sta dietro la DE-INTENSIFICAZIONE della ART è di eliminare della terapia antiretrovirale, in modo strutturato e sicuro, per arrivare a capire qual è la fonte della recrudescenza virale e quale la natura dei reservoir. Di qui alcuni studi che verranno presentati in questi giorni.
Lo studio di Schacker dimostra che , dopo interruzione del trattamento, si ha un rebound virale nei tessuti linfatici, simultaneamente in diversi comparti anatomici.
Questo può dipendere o dalla presenza di un grande serbatoio di cellule latentemente infette – e allora ci si deve chiedere come abbiano fatto ad attivarsi tutte insieme in diversi siti anatomici.
Oppure (spiegazione alternativa, ma non necessariamente incompatibile) da cellule che sono infette non in modo latente, ma produttivo, che non sono in grado di causare nuove infezione finché subiscono la pressione della ART ma, quando questa viene sospesa, possono innescare nuovi cicli di infezione.
Passiamo ora alle diverse strategie che la ricerca sta mettendo in atto per arrivare all’eradicazione.
Una strategia che è già passata in fase clinica è lo “shock and kill” – il tentativo di ripulire il reservoir attivando il virus dalla latenza.
Attivatori della latenza dell’HIV.
Il farmaco antilatenza che è stato ad oggi più studiato è il Vorinostat, con risultati per ora assai ambigui. [NdD: è vero che lo studio portato al congresso dalla Lewin riguarda soprattutto la sicurezza della somministrazione di questo HDACi ma … che delusione!!]
Le aspettative di qualsiasi strategia di “purging” dei reservoir sono che la riattivazione del virus porti all’eliminazione delle cellule infette, sia grazie agli aspetti citopatici del virus, sia grazie alla reazione immune dell’ospite.
Ma la strada è resa più difficile dal fatto che Siliciano ha dimostrato che la frequenza di provirus capaci di replicazione è più alta di quanto si pensasse.
C’è poi la questione dei reservoir nelle cellule della linea mieloide, e segnatamente i macrofagi, che potrebbero – se si dimostrassero davvero un reservoir presente in persone con viremia soppressa dalla ART - richiedere delle strategie di eradicazione diverse da quelle che si stanno sperimentando contro cellule della linea linfoide.
La ricerca che sta facendo Stevens riguarda proprio l’uso di Vorinostat, acido valproico e Disulfiram su linee cellulari di macrofagi.
In conclusione, le questioni principali che rimangono aperte riguardano
- 1) il contributo dell’infezione residua alla persistenza virale e i suoi reali effetti clinci;
2) l’origine del rebound virale quando si sospende la ART;
3) il problema se la cronicità dell’HIV sia limitata ai linfociti T latentemente infetti e se questo richieda strategie di purging differenti;
4) il dubbio che i farmaci antilatenza non siano in grado di accelerare la distruzione delle cellule latentemente infette;
5) la difficoltà di trovare i test più adatti per misurare i progressi dei trial clinici di cura dell’infezione.
Alla fine della lezione, rispondendo a una domanda sulle prospettive dei farmaci antilatenza da qui a 5 anni, Stevenson ha risposto che ha dei grossi dubbi sui test che abbiamo oggi a disposizione per studiare gli effetti di queste sostanze sulla latenza virale. Il gold standard continua ad essere la rilevazione di virus capace di replicazione, ma i test oggi correnti mancano di precisione e danno risultati che non si riescono a riprodurre da un laboratorio all'altro [cosa che è emersa in modo INCREDIBILE l'estate scorsa, quando non si capiva se fosse stato trovato o no del virus capace di replicarsi in Timothy Brown].
Altra domanda: come combinare le diverse strategie per attaccare i reservoir?
Risposta: unire a una strategia antilatenza la soluzione del "problema di Shen-Siliciano" e fare in modo di rinforzare gli effetti citotossici dei CD8 [vabbè, questo l'avevamo capito anche noi fin dall'anno scorso ...]
